Posted onInUni-MolWord count in article: 1.1kReading time ≈4 mins.
Pre-trained models are sweeping through the AI field by extracting representative information from large-scale unlabeled data and then performing supervised learning on small-scale labeled downstream tasks, becoming the de facto solution in many application scenarios. In drug design, there is still no consensus on the "best way to represent molecules." In the field of materials chemistry, predicting molecular properties is equally important. Mainstream molecular pre-training models typically start from one-dimensional sequences or two-dimensional graph structures, but molecular structures are inherently represented in three-dimensional space. Therefore, directly constructing pre-trained models from three-dimensional information to achieve better molecular representations has become an important and meaningful problem. To further promote research on molecular representation and pre-trained models, Uni-Mol will join the DeepModeling community to work with community developers to advance the development of a three-dimensional molecular representation pre-training framework.
Posted onInOpenLAMWord count in article: 721Reading time ≈3 mins.
On the journey toward developing a Large Atomic Model (LAM), the core Deep Potential development team has launched the OpenLAM initiative for the community. OpenLAM’s slogan is "Conquer the Periodic Table!" The project aims to create an open-source ecosystem centered on microscale large models, providing new infrastructure for microscopic scientific research and driving transformative advancements in microscale industrial design across fields such as materials, energy, and biopharmaceuticals.
Posted onInDflowWord count in article: 733Reading time ≈3 mins.
From the development of software ecosystems in fields such as electronic structure calculations and molecular dynamics, to the systematic evaluation of large models like OpenLAM, and gradually addressing scientific and industrial R&D problems such as biological simulations, drug design, and molecular property prediction, a series of AI4Science scientific computing software and models are rapidly advancing. This progress is closely linked to better research infrastructure, with the Dflow project being a key component.
Posted onInOpenLAMWord count in article: 711Reading time ≈3 mins.
The slogan for OpenLAM is "Conquer the Periodic Table!" We hope to provide a new infrastructure for microscale scientific research and drive the transformation of microscale industrial design in fields such as materials, energy, and biopharmaceuticals by establishing an open-source ecosystem around large microscale models. Relevant models, data, and workflows will be consolidated around the AIS Square; related software development will take place in the DeepModeling open-source community. At the same time, we welcome open interaction from different communities in model development, data sharing, evaluation, and testing.
Posted onInJAX-FEMWord count in article: 761Reading time ≈3 mins.
"The integration of machine learning and physical modeling is revolutionizing the paradigm of scientific research. People aiming to push the boundaries of science and solve challenging problems through computational modeling are coming together in unprecedented ways." Recently, the DeepModeling open-source community has welcomed a new member in the field of macro-scale computation. To further advance the development of the JAX-FEM project, a differentiable finite element method library, JAX-FEM will join the DeepModeling community. Together with developers and users in the community, it aims to expand the frontiers of finite element methods in the AI4Science era.
Community project homepage: https://github.com/deepmodeling/jax-fem
Posted onInOpenLAMWord count in article: 1.7kReading time ≈6 mins.
The slogan for OpenLAM is "Conquer the Periodic Table!" We hope to provide a new infrastructure for microscale scientific research and drive the transformation of microscale industrial design in fields such as materials, energy, and biopharmaceuticals by establishing an open-source ecosystem around large microscale models. Relevant models, data, and workflows will be consolidated around the AIS Square; related software development will take place in the DeepModeling open-source community. At the same time, we welcome open interaction from different communities in model development, data sharing, evaluation, and testing.
Posted onInOpenLAMWord count in article: 626Reading time ≈2 mins.
Peter Thiel once said, "We wanted flying cars, instead we got 140 characters (Twitter)." Over the past decade, we have made great strides at the bit level (internet), but progress at the atomic level (cutting-edge technology) has been relatively slow.
The accumulation of linguistic data has propelled the development of machine learning and ultimately led to the emergence of Large Language Models (LLMs). With the push from AI, progress at the atomic level is also accelerating. Methods like Deep Potential, by learning quantum mechanical data, have increased the space-time scale of microscopic simulations by several orders of magnitude and have made significant progress in fields like drug design, material design, and chemical engineering.
The accumulation of quantum mechanical data is gradually covering the entire periodic table, and the Deep Potential team has also begun the practice of the DPA pre-training model. Analogous to the progress of LLMs, we are on the eve of the emergence of a general Large Atom Model (LAM). At the same time, we believe that open-source and openness will play an increasingly important role in the development of LAM.
Posted onInDeepSPINWord count in article: 1.5kReading time ≈5 mins.
In the field of magnetic material science, many frontier challenges require considering the coupling between lattices and spins at the atomic scale. For example, areas such as ultrafast magnetization dynamics, terahertz spintronics, and magnetocaloric materials rely on simulations of energy transfer between lattice and spin subsystems. Additionally, the interplay between magnetism and the lattice directly impacts the performance of materials critical to high-voltage power transmission, the automotive industry, and high-performance batteries.
To address these challenges, researchers have employed various multi-scale simulation methods, such as first-principles calculations, spin dynamics, and micromagnetics. However, these methods exhibit significant limitations when describing material defects, polycrystalline and disordered materials, finite-temperature-related properties, and magnetic dynamic processes.
To overcome these obstacles, the research group led by Xu Ben at the Graduate School of China Academy of Engineering Physics (GSCAEP) has developed an atomic-scale lattice-spin potential generation method called DeepSPIN. This tool supports atomic simulations of magnetic materials in the AI for Science era, enabling the exploration of uncharted territories in the world of magnetic materials.
1. DeepSPIN: High-Precision Description of Complex Lattice-Magnetic Coupling
The research group led by Ben Xu at the Graduate School of China Academy of Engineering Physics (GSCAEP) has developed DeepSPIN, a method for generating atomic-scale lattice-spin potential energy models. DeepSPIN aims to accurately predict the energy, atomic forces, and magnetic torques of systems under simultaneous lattice and spin perturbations. With DeepSPIN, researchers can simulate the equilibrium and excited-state configurations of lattices and spins and describe their evolution processes. This model, focusing on atomic-scale potential energy generation for non-collinear magnetic configurations, holds significant importance in addressing many challenges in the field of magnetic material science. DeepSPIN's functionalities were officially released in June 2023.
Key Innovations:
Pseudo-atomic Approach: DeepSPIN employs a novel "pseudo-atomic" method to accurately describe the relative orientations between spins and lattices.
Integration with AI: The method combines deep learning and active learning strategies to capture multi-degree-of-freedom landscapes at varying energy scales.
First-Principles Data: Training data is generated using the DeltaSPIN magnetic constraint method, developed by the same research group.
Development and Collaboration: DeepSPIN was initiated by Ben Xu (GSCAEP), Han Wang(Institute of Applied Physics and Computational Mathematics, Beijing), and Linfeng Zhang (Beijing Academy of Artificial Intelligence), with Ben Xu leading development efforts. AI modeling support was provided by Teng Yang (Tsinghua University), Zhengtao Huang (Wuhan University of Technology), and Duo Zhang (DP Technology). The DeepModeling community supported code hosting and technical development.
1.1 Challenges Addressed by DeepSPIN
When simultaneously considering spin and lattice subsystems, it is necessary to address two dominant equations:
When considering both spin and lattice subsystems simultaneously, two main governing equations need to be addressed:
Unlike a pure lattice or pure spin system, the atomic force (\vec{F}) here is influenced by the magnetic structure, and the spin precession matrix (\vec{\omega}) is affected by the lattice structure. DeepSPIN can determine the total energy (E) of the system and provide the (\vec{F}) and (\vec{\omega}) for each atom based on different lattice and magnetic configurations.
The DeepSPIN project aims to address the following challenges:
Incorporating Magnetic Ordering in Atomic-Scale Simulations How to account for the influence of magnetic ordering on phase transitions, transport processes, and other phenomena in existing atomic-scale simulation methods.
Describing the Coupling Between Coordinates and Spins How to accurately represent the mutual coupling between atomic positions and spins in atomic potential energy functions.
Predicting Spin Evolution How to predict the evolution of spin orientations starting from atomic structures and magnetic configurations.
Modeling Non-Trivial Magnetic Configurations How to accurately describe non-trivial magnetic configurations, such as those found at defects and interfaces in complex crystal structures.
Capturing Realistic Magnetic Configurations at Different Temperatures How to accurately represent magnetic configurations at various temperatures, including paramagnetic states and spin fluctuations.
Simulating Lattice and Spin Dynamics in Magnetic Materials How to precisely describe the dynamic processes of lattices and spins in magnetic materials.
1.2 Current Capabilities of DeepSPIN
By integrating with the existing LAMMPS (Spin) framework, DeepSPIN can achieve the following:
Prediction of Key Properties
Predict the energy, atomic forces, and magnetic torque for complex crystal structures and non-collinear magnetic configurations.
Ground-State Configurations
Determine the ground-state configurations of both atomic and magnetic systems through iterative energy minimization of the lattice and spin subsystems.
Thermodynamic Descriptions
Describe magnetic behavior at finite temperatures under thermodynamic ensembles.
Applicable Systems
Supports simulations of ferromagnetic and antiferromagnetic materials in both metals and insulators. Current model materials include:
Fe (Iron)
NiO (Nickel Oxide)
BiFeO(_3) (Bismuth Ferrite)
1.3 Accuracy of DeepSPIN
Energy Prediction Accuracy: Generally reaches a precision of approximately (10^{-4}) eV.
Magnetic Torque Prediction Accuracy: Achieves a precision of about 10 meV/(\mu_B).
This level of precision makes DeepSPIN highly reliable for exploring complex magnetic material systems.
2. Cases Examples
Potential Energy Surface of NiO
Taking the simple antiferromagnetic material NiO as an example, three typical energy-scale events were selected:
Atomic Displacement (D): Movement of atoms from their equilibrium positions.
Relative Spin Rotation (C): Rotation of neighboring spins relative to each other.
Collective Spin Rotation (R): Simultaneous rotation of neighboring spins in the same direction.
The high-dimensional potential energy surface of NiO was projected into a 3D space defined by these events. This example demonstrates that DeepSPIN can accurately explore the potential energy surface of materials governed by complex energy-scale interactions.
Magnetic Configurations at NiO Grain Boundaries
Grain boundaries play a critical role in determining the functionality of magnetic material devices. However, the complex lattice structures at these boundaries greatly influence the magnetic properties of the device. DeepSPIN effectively predicts magnetic configurations and interactions at such complex structures (e.g., grain boundaries), providing valuable insights for optimizing magnetic devices.
For NiO, a classical antiferromagnetic material, grain boundaries are common atomic-scale defects. A specific symmetric grain boundary structure was analyzed, comprising 11,520 atoms with mirror symmetry across the boundary. At the grain boundary, atomic displacements deviate from the equilibrium configuration of a perfect crystal. For the first time, DeepSPIN revealed the magnetic configurations at such grain boundaries, offering a novel perspective for studying their impact on material properties.
Magnetic Switching Barrier in BiFeO(_3)
BiFeO(_3) (Bismuth Ferrite) is a representative multiferroic material with two order parameters:
Polarization (P): Related to structural distortions of the Fe-O octahedra.
Magnetization (S): Closely associated with the lattice distortions.
In BiFeO(_3), the polarization (P) can be reversed under an external field. DeepSPIN was used to simulate one such reversal pathway, showing:
Polarization Change: P gradually varies in magnitude during the reversal.
Magnetization Stability: S remains constant in both direction and magnitude.
Atomic Displacement: Reversal involves significant atomic displacements of up to 0.5 Å.
DeepSPIN demonstrated excellent performance in scenarios where the relative orientations of lattice and magnetic moments undergo significant changes, providing accurate predictions of energy barriers and dynamics.
3. Future Development Plans for DeepSPIN
DeepSPIN will focus on addressing the following challenges in its future development:
Effects of Random Doping
Developing methods to describe how random doping impacts the magnetic configurations of materials.
Transition-State Simulations in Coupled Systems
Modeling the transition states during phase transitions in systems with coupled lattice and magnetic properties.
Phase Transitions Under External Fields
Accurately describing the phase transition processes (both lattice and magnetic moments) under external fields such as force fields, temperature fields, and magnetic fields.
Time-Dependent Evolution of Lattice and Spin Configurations
Realizing time-dependent simulations of lattice and spin configurations, including high-performance optimization within the LAMMPS framework.
4. DeepSPIN Project Repository
The DeepSPIN project has been fully integrated into DeePMD-kit version 2.2.2 and is available for use in version 2.2.2 or later. You can access the project at the following link: DeepMD-kit GitHub Repository
Posted onInAPEXWord count in article: 663Reading time ≈2 mins.
The AI for Science approach, exemplified by DeePMD-kit, has demonstrated immense potential in precise molecular/atomic-scale simulations. At the same time, a robust, efficient, and automated model evaluation system plays a vital role in the ongoing development and practical applications of these methods.
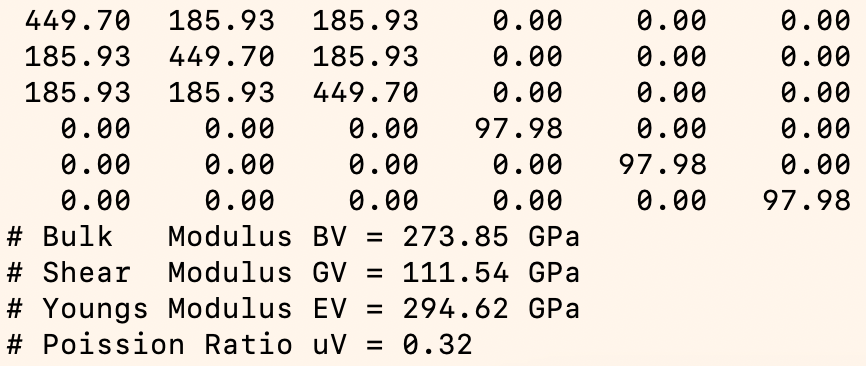
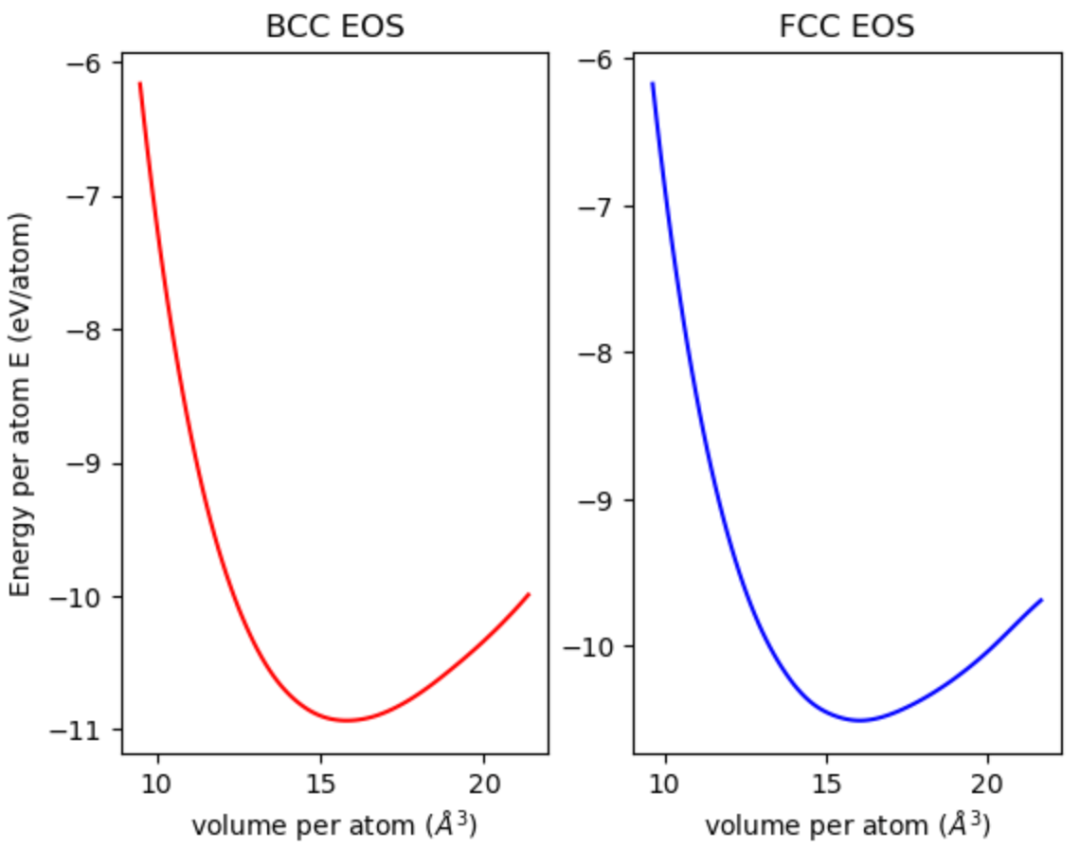
For alloy systems, researchers are particularly interested in the calculation of equilibrium structures and energies, energy-volume relationships, elastic constants, vacancy formation energy, interstitial defect formation energy, and surface formation energy. Comparing these properties with Density Functional Theory (DFT) or experimental results for the same alloy system helps evaluate model accuracy. For machine learning potentials like Deep Potential (DP), such evaluations can also guide optimization of training strategies and parameter settings during preliminary training.
To address these needs, developers from the DeepModeling community, including @kevinwenminion and @ZLI-afk, leveraged the capabilities of the maturing dflow cloud-native workflow for scientific computing. They released an open-source software package called APEX (Alloy Properties EXplorer using simulations). The project repository is available at: APEX GitHub.
1 Historical Background:
The early workflow for alloy property calculations was released in 2019 as part of the DP-GEN code and functioned as a submodule, dpgen autotest. Despite multiple optimizations and iterations, this setup faced challenges:
Significant redundancy in development efforts when adding new property calculation modules or simulation methods, making maintenance difficult.
Weak error detection and exception handling capabilities.
In response, developers rebuilt the alloy property workflow using dflow's comprehensive and user-friendly workflow development tools, along with tools like the first-principles operator library fpop. The restructured workflow was named Alloy Properties EXplorer using simulations (APEX). APEX aims to provide the community with an efficient, user-friendly, and highly compatible workflow for alloy property testing.
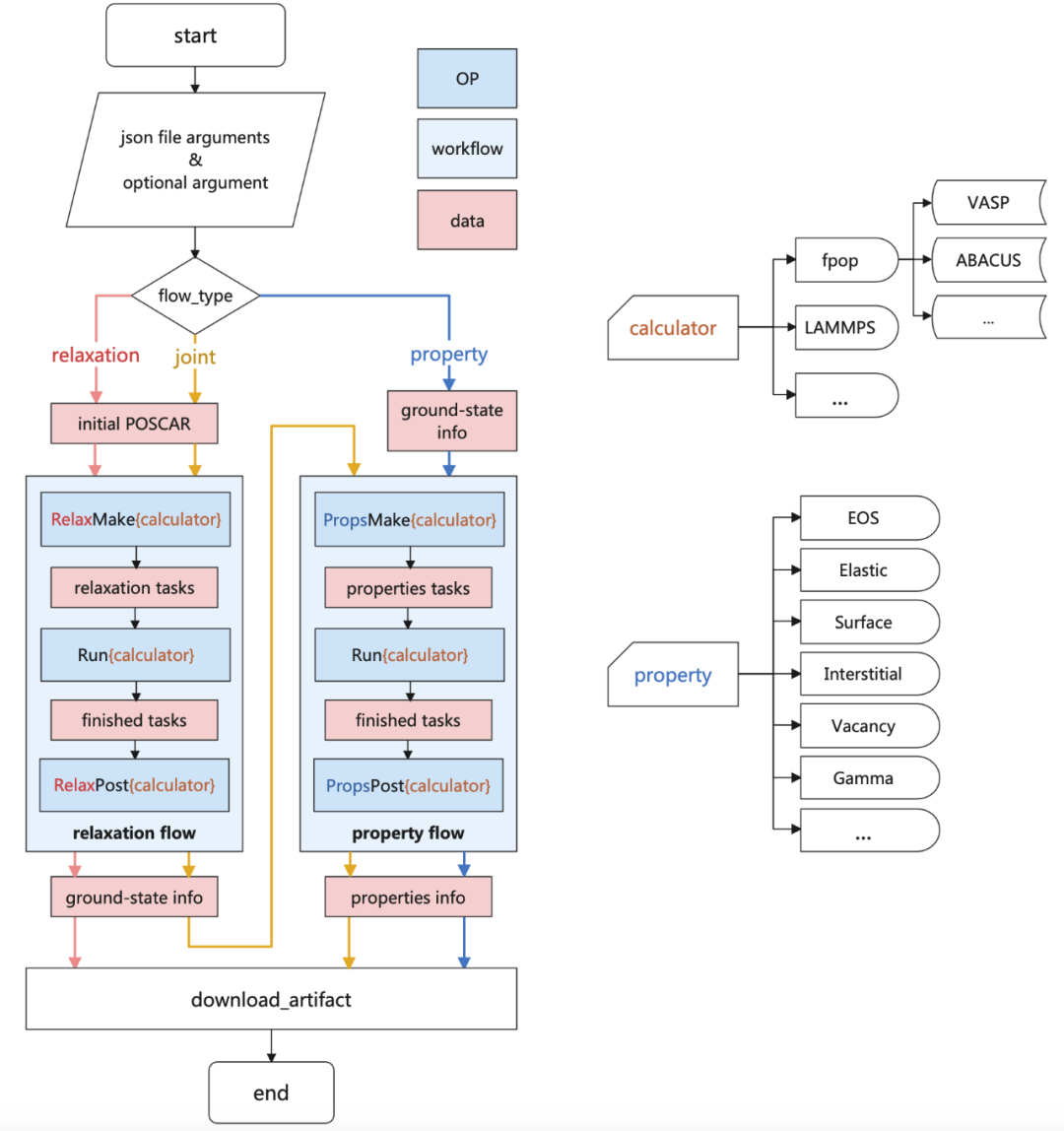
2 Current Features of APEX
The current APEX alloy property testing workflow supports:
First-principles calculations (VASP, ABACUS)
Molecular dynamics calculations (LAMMPS)
Currently supported alloy properties include:
Energy-volume relationship curves (EOS)
Elastic constants
Surface energy
Interstitial formation energy
Vacancy formation energy
Stacking fault energy (Gamma Line)
New property calculation functionalities will be added in future updates.
2.1 Key Highlights of APEX:
Efficiency in Workflow Management: Enhanced by the integration with dflow, ensuring smooth process control.
Simplicity and User-Friendliness: Easy to install and operate, with intuitive interfaces and intelligent interactions.
Parallel Task Execution: Enables parallel submission of multiple configurations and properties, with one-click execution and result retrieval.
High Extensibility: Facilitates expansion to support additional properties and computational software.
2.2 APEX Workflow Overview
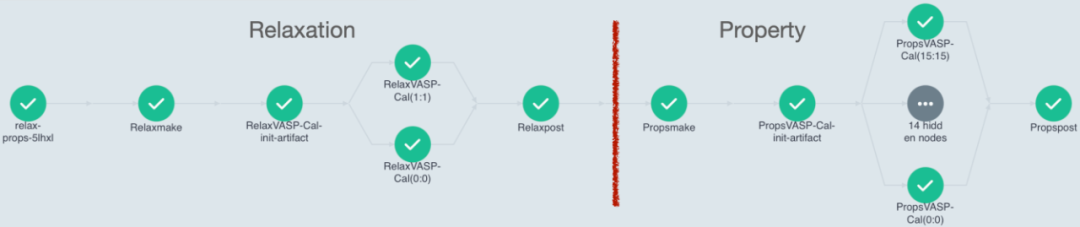
APEX maintains a two-step structure for alloy property calculations:
Relaxation
Property Calculation
These steps are encapsulated into two independent workflows, allowing users to execute either step independently. Alternatively, APEX provides a joint workflow that merges both steps for a streamlined, one-click property testing process.
2.3 Example Outputs:
Full Workflow for Relaxation + Testing (Joint Workflow)
Elastic Constants and Modulus
Equation of State (EOS)
By combining advanced AI methods, user-friendly design, and open collaboration, APEX establishes itself as a powerful tool for alloy property exploration in scientific research and engineering applications.
The full code for APEX is available on GitHub at https://github.com/deepmodeling/APEX.
Looking ahead, APEX will expand to support functionalities such as phonon spectrum calculations, dislocation structure calculations, and finite-temperature property calculations. It will also enhance post-processing capabilities to enable automatic extraction of results, plotting, and preparation of final reports, making it easier for users to intuitively analyze test results.
We welcome everyone to provide feedback by submitting issues on APEX's GitHub repository or contribute code via pull requests.